Medical (Doctor Talk)
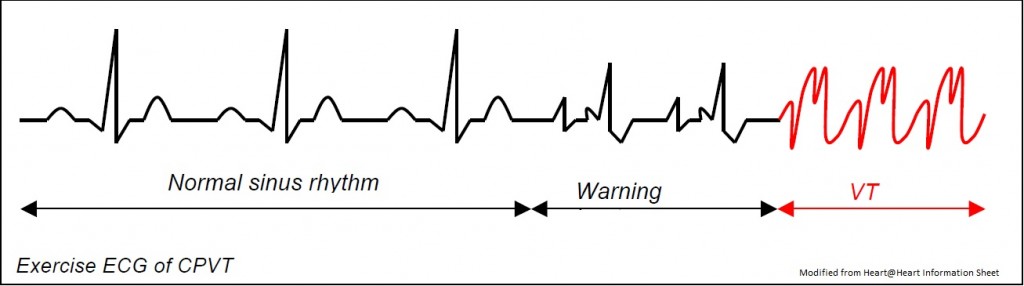
CPVT is a rare arrhythmogenic disease characterised by exercise or stress induced ventricular arrhythmias, syncope or sudden death, usually in the paediatric age group, including infancy [Tester 2004, Francis 2005]. Familial occurrence has been noted in at least a third of cases, and inheritance can be dominant or recessive, with high penetrance. Like LQTS, CPVT can be mistaken for epilepsy and be treated, inappropriately, with long-term anticonvulsants. Moreover, similar to LQTS type 1, swimming with CPVT can trigger arrhythmogenic events, and is suspected as an important cause of unexpected drowning [Ackerman 1998, Moss 1999, Choi 2004]. The resting ECG is normal. Exercise test, Holters or digital loop recorders may reveal polymorphic ventricular tachycardia during exercise (Coumel 2002), though this is not completely reliable. The mainstay of treatment is beta blockade, which certainly reduces the episodes of VT on Holter (Coumel 2002). However on follow up of patients receiving beta blockade over 7 years, one group found a mortality of 10% [Leenhardt 1995], whilst another found 25% [Sumitomo 2003]. Missing a single dose of beta blocker can be fatal, and defibrillator pacemakers are being used more frequently. It is now known that Flecainide is extremely beneficial in cases unresponsive to beta blockers, and is often used in addition.
Genetics
Mutations within the cardiac Ryanodine gene (RyR2) have been identified in autosomal dominant pedigrees [Priori 2001, Laitinen 2001]. Three different isoforms have been identified, however, it is ryanodine receptor type 2 (RyR2) that is expressed in cardiac muscle. RyR1 is the skeletal muscle ryanodine receptor that is involved in malignant hyperthermia [Tiso 2001]. The receptors share a common molecular structure, whereby channels are formed as homotetramers composed of four RyR2 polypeptides. The Ca2+ channels regulate excitation-contraction coupling in cardiac muscle. Normally, the receptor is regulated by a range of accessory proteins resulting in normal Ca2+ homeostasis. The RyR2 is a large gene, made up of 105 exons, but mutations are clustered within three distinct regions of the gene, covering 40 exons [tester 2004]. Point mutations within RyR2 account for CPVT in at least 38% of cases [Laitinen 2001]. The mechanisms by which mutations in the cardiac ryanodine receptor sometimes lead to catecholamine-mediated ventricular tachycardia and sometimes to ARVC are not clear. CASQ2- a calcium binding protein located in the sarcoplasmic reticulum – has also been implicated in a severe and uncommon autosomal recessive form of CPVT, [Postma 2002, and Lahat 2001].
In a screening study of DNA from 49 victims of sudden death with a negative post-mortem, with a mean age of 14 years, revealed that 7 (14%) had pathological mutations within RyR2 [Tester 2004]. The youngest child was aged 2 years; he died suddenly whilst watching TV. Mutations are also found very rarely in SIDS victims.
Screening for asymptomatic gene carriers is hampered by the fact that the resting ECG is normal. Exercise testing and Holter monitoring can be used, but the interpretation of the result in this context is difficult, particularly if there is a negative result. Compared to LQTS “the lethality associated with CPVT seems greater and it may represent the ideal arrhythmogenic killer that is able to escape suspicion, detection, and apprehension by either a standard medico-legal autopsy or a careful evaluation of surviving first and second degree relatives” [Tester 2004]. It would thus be very desirable to be able to offer genetic testing when the post mortem from a sudden death is negative, to facilitate screening within such families.
Management
This involves use of high dose beta blockers, left cardiac sympathectomy, flecainide and in some cases, defibrillator pacemakers. Subjects should avoid swimming and competitive sports, at least until repeat exercise testing on treatment confirms absence of arrhythmias.